- Braquimetatarsia
- Deformidades do 5º dedo
- Sindactilia e Polidactilia
- Gigantismo e Macrodactilia
- Pé Fendido
- Charcot-Marie-Tooth
- Pé Torto Congênito
Braquimetatarsia
Os metatarsos são ossos longos e formam o prolongamento anterior do pé, articulam-se isoladamente com cada um dos cinco dedos na sua porção mais distal (ponta do pé) e com quatro ossos do tarso (cuneiforme medial, cuneiforme intermédio, cuneiforme lateral e cubóide).
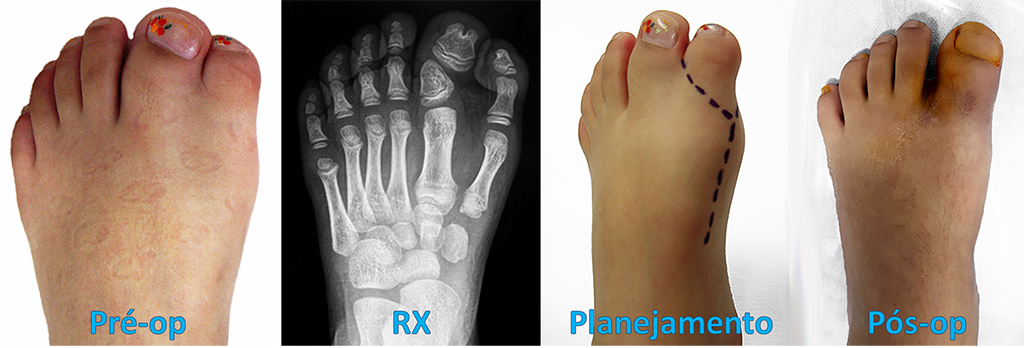
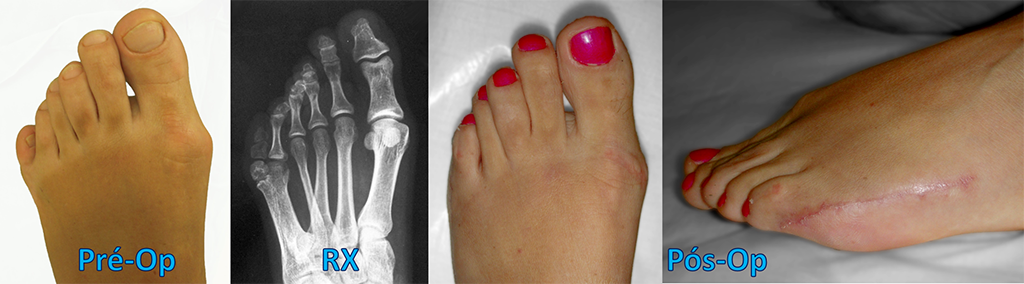
Braquimetatarsia quer dizer “ metatarso curto ” (do grego Brachys = curto). É uma alteração congênita, frequentemente bilateral (72%) e mais prevalente no sexo feminino (25:1).
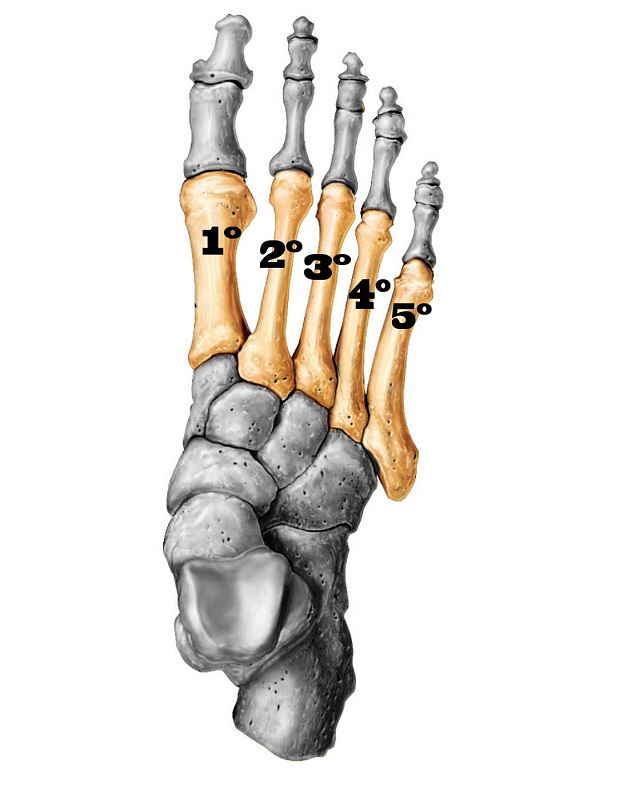
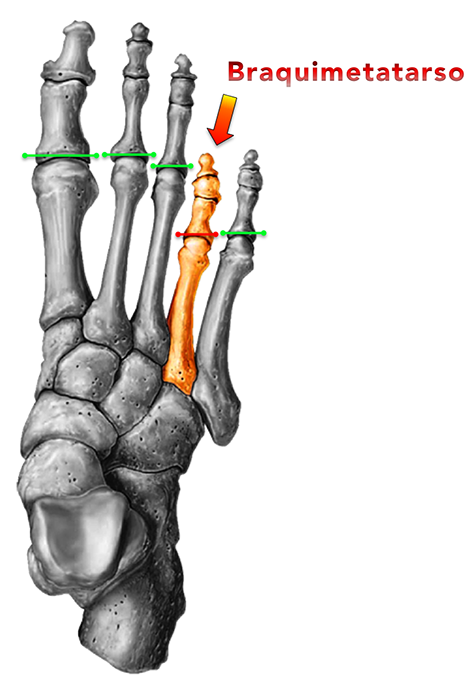
A braquimetatarsia congênita é uma hipoplasia (diminuição da formação) ocasionada pelo fechamento precoce da cartilagem de crescimento (fise) e envolve principalmente o primeiro, o terceiro e/ou o quarto metatarsos. Isso restringe o desenvolvimento ósseo na infância e torna o metatarso mais curto em relação aos demais na vida adulta.
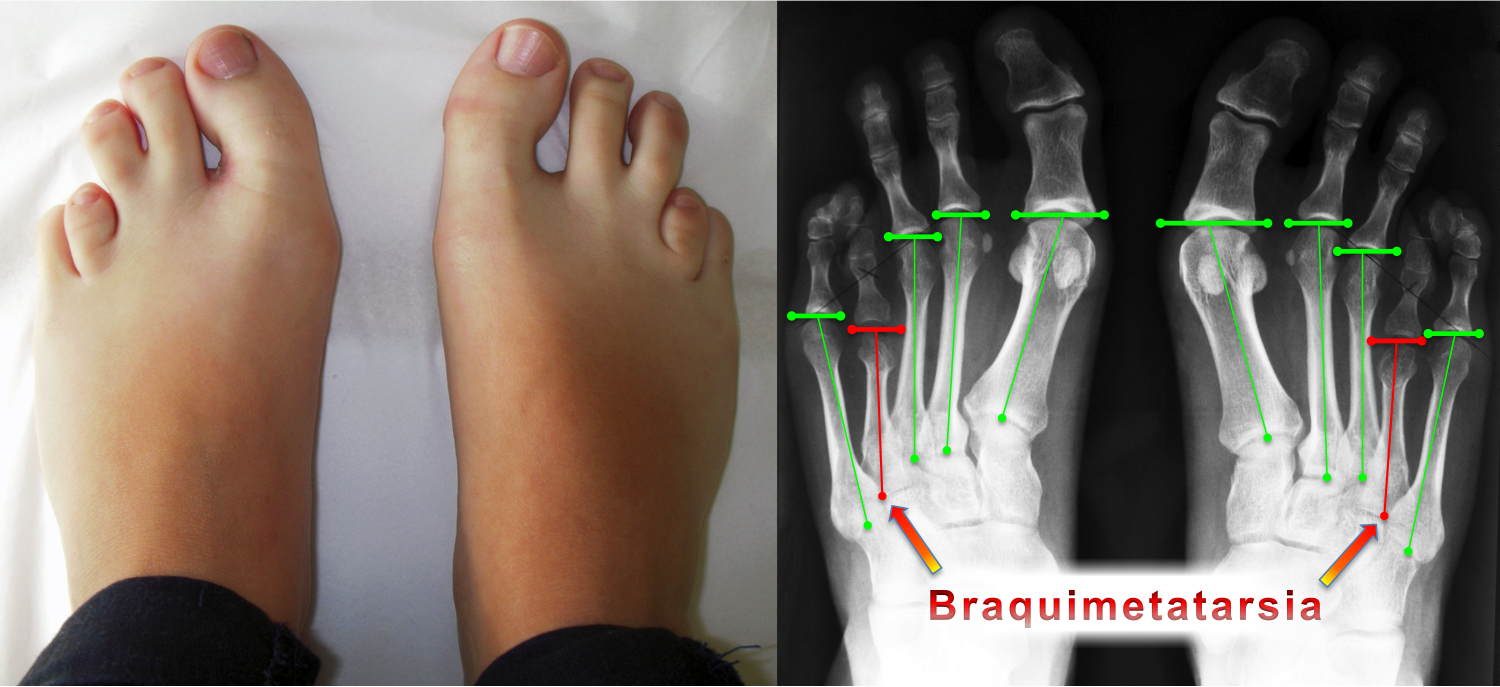
A fórmula metatarsal (estudada por Viladot, Hardy e Clapham) é a linha de relação entre as cabeças dos metatarsos e sua protusão anterior. A braquimetatarsia altera essa fórmula metatarsal e, consequentemente, a distribuição da carga de apoio na região frontal do pé.
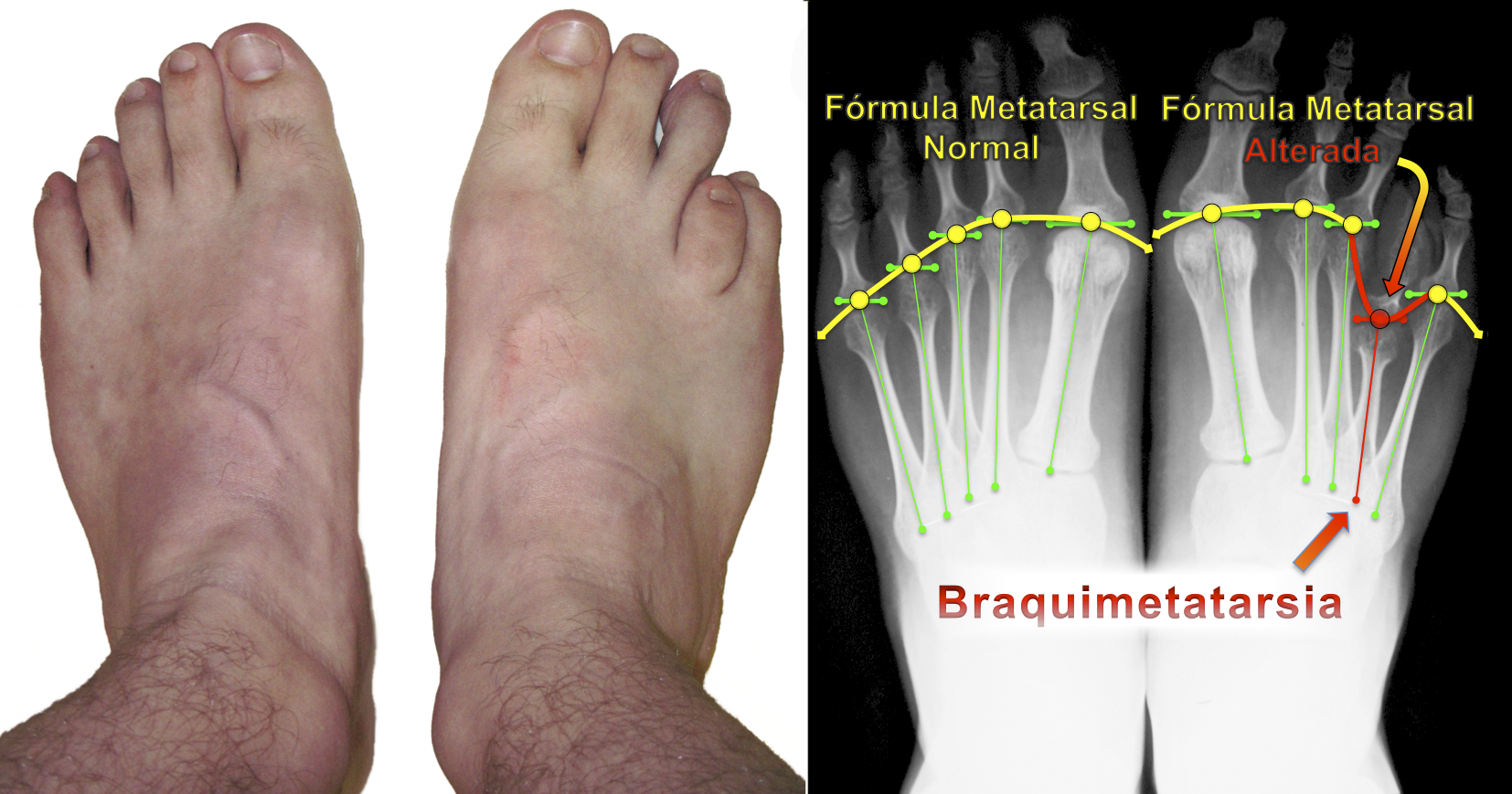
Dor e calosidades plantares podem estar presentes pela má distribuição da pressão na frente do pé. Também pode ocorrer a elevação do dedo relacionado ao metatarso encurtado, causando desconforto pelo contato dorsal com o calçado.
Fatores hereditários da deformidade podem estar presentes em algumas famílias ou associados à síndrome de Down, síndrome de Turner, síndrome de Apert ou osteodistrofia de Albright. A braquimetatarsia também pode ocorrer em consequência de traumas, infecções, tumores ou outros acometimentos que resultam no encurtamento metatarsal.
Pacientes com encurtamento metatarsal congênito quase sempre não apresentam queixas importantes de dor ou incapacidade funcional. A principal queixa portanto é quanto a forma estética do pé, o que leva a constrangimentos ao usar calçados abertos, chinelos ou andar com os pés descalços.
A avaliação da braquimetatarsia é feita através de exames de RX de ambos os pés com apoio e, de forma complementar, com tomografia computadorizada.
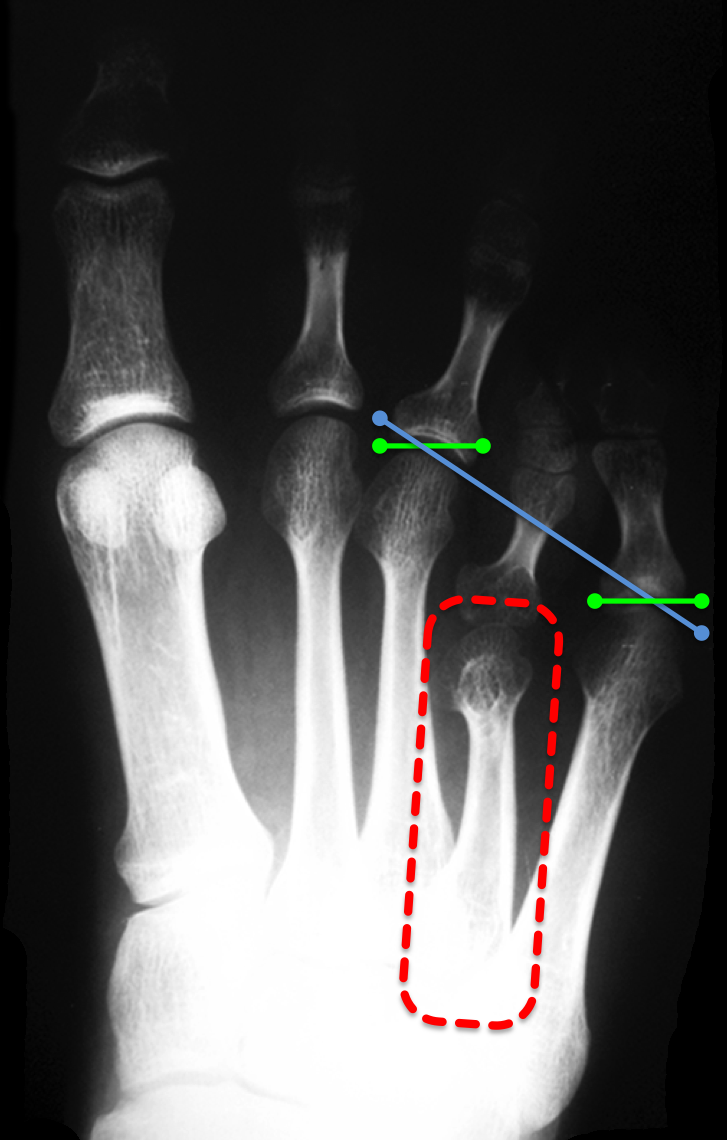
O tratamento baseia-se nas queixas apresentadas pelo paciente e pode ser feito de forma conservadora ou cirúrgica.
O tratamento conservador tem por objetivo distribuir uniformemente a pressão plantar metatarsal e diminuir o contato sobre as proeminências ósseas do pé, minimizando a dor e as calosidades presentes. Para isso, pode-se orientar a mudança do tipo de calçado e adaptar palmilhas moldadas visando o conforto.
O tratamento cirúrgico tem por objetivo alongar o osso metatarsal encurtado, melhorando o equilíbrio e a distribuição de carga plantar, além de promover a satisfação do paciente pela restauração estética do pé.
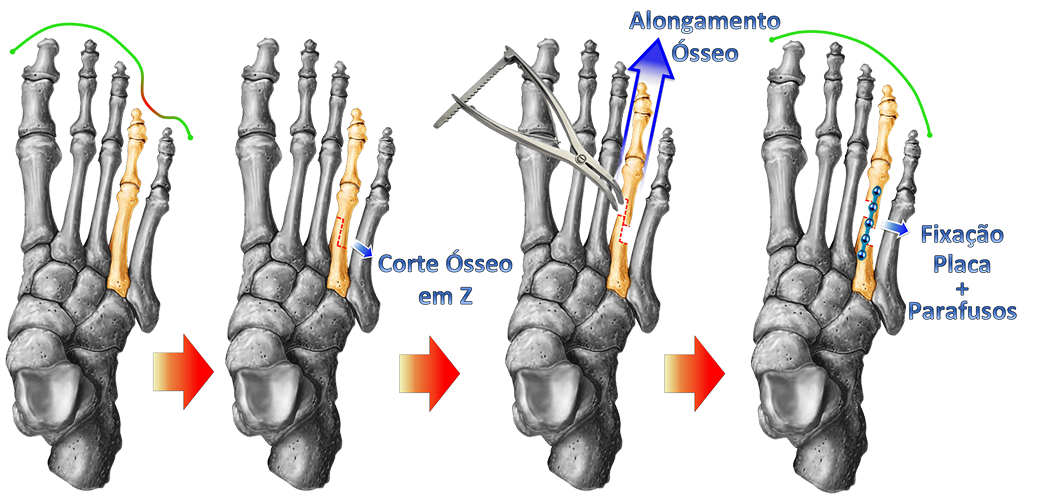
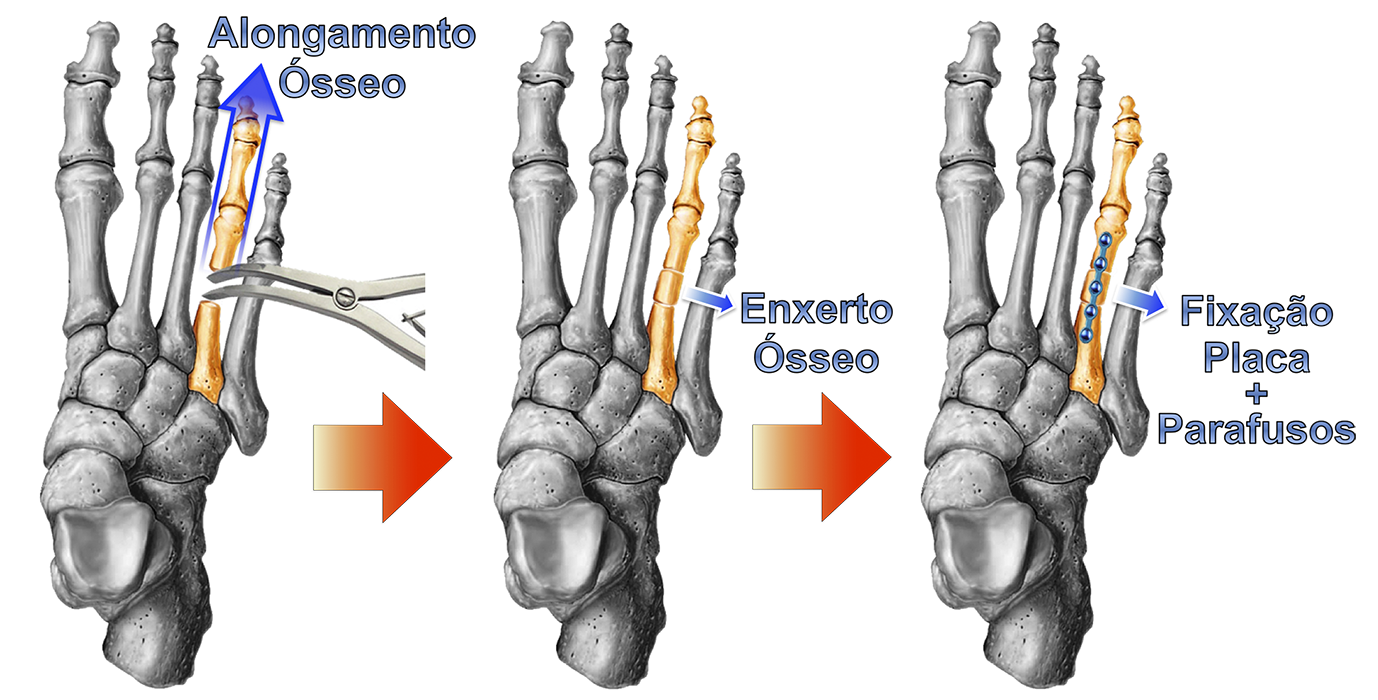
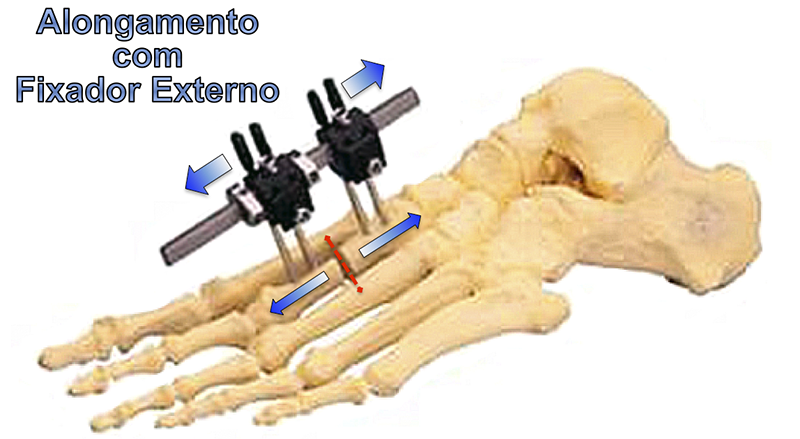
Existem várias técnicas cirúrgicas descritas na literatura médica para a correção da braquimetatarsia. As mais utilizadas são os alongamentos, com ou sem enxerto ósseo, concomitante ou não com o encurtamento de outros metatarsos, e o uso de fixadores externos.
ALONGAMENTO IMEDIATO COM OU SEM USO DE ENXERTO ÓSSEO :
ALONGAMENTO PROGRESSIVO COM USO DE FIXADOR EXTERNO :


Deformidades do 5º dedo
Sobreposição e Sobposição do 5º Dedo
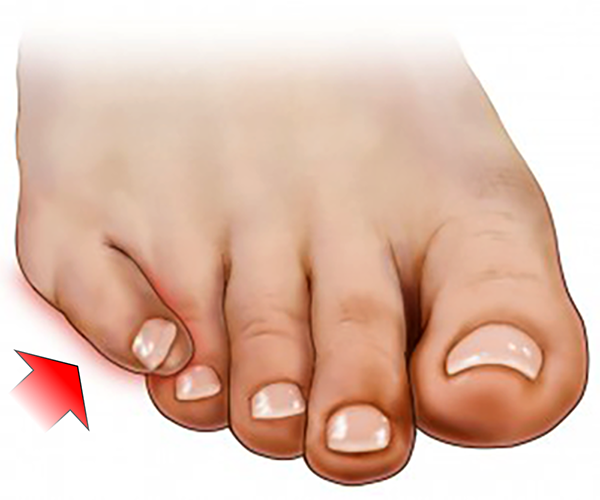
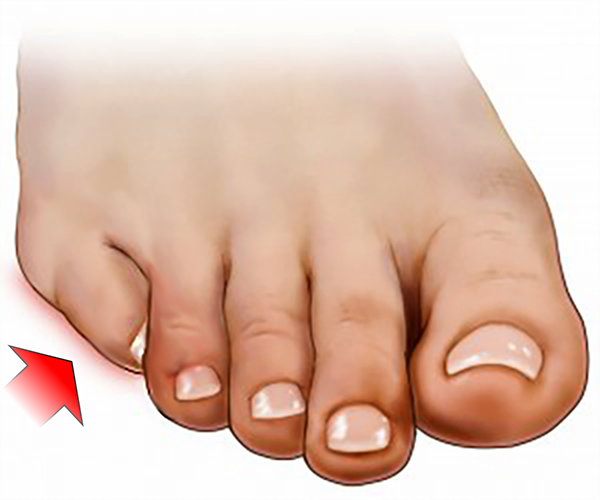
SOBREPOSIÇÃO SOBPOSIÇÃO
Sobreposição do 5º Dedo (Overllaping ou Supra-Aduto)
A sobreposição do quinto dedo do pé (menor dedo ou “dedinho” do pé) é uma deformidade congênita muito comum na população.
Também pode ser descrita como Overllapping ou 5º dedo Supra-Aduto, isto é, desviado para cima e para dentro.
Caracteriza-se pela rotação e desvio medial das falanges do 5º dedo e contratura do tendão extensor longo, assumindo um posicionamento por cima do 4º dedo.
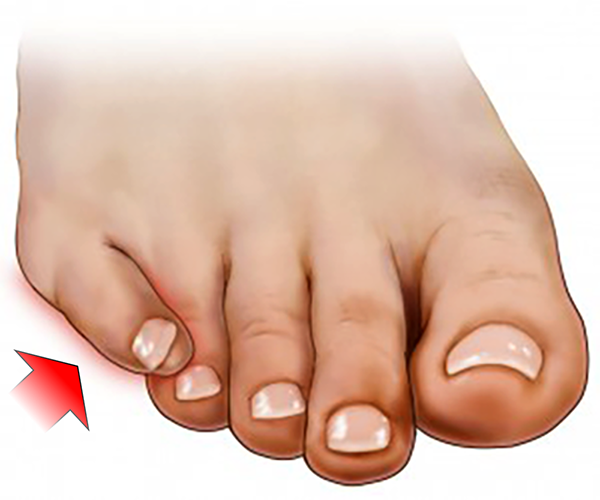
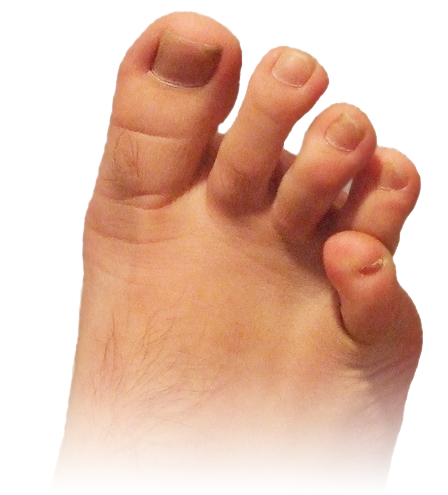
Essa deformidade causa bastante incômodo, principalmente nas mulheres, pelo uso de calçados de salto alto e bico fino, que comprimem e fazem atrito lateral no pé.
Muitas vezes ocorre a formação de uma calosidade na porção mais lateral e superior do dedo ou entre o 4º e o 5º dedos (calo em espelho). A unha pode sofrer importante alteração na sua forma e no crescimento.
O tratamento conservador baseia-se na proteção com coxins (pads) para acolchoamento e mudança do estilo de calçados.
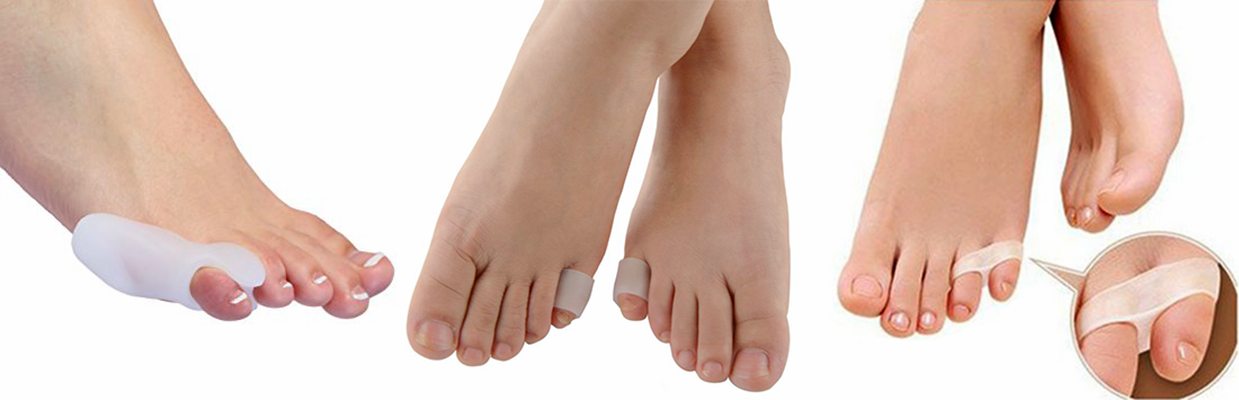
O tratamento cirúrgico para os casos leve e moderados consiste no alongamento e liberação das estruturas que estão encurtadas e contraturadas, como o tendão extensor longo e a cápsula articular.
O tratamento cirúrgico para deformidades mais severas consiste em uma plastia da pele, isto é, a criação de um retalho em V-Y para alongamento do tecido cutâneo, juntamente com o alongamento do tendão extensor longo e da cápsula articular.
Sobposição do 5º dedo (Underllaping ou Infra-Aduto)
Assim como a sobreposição, a sobposição do quinto dedo do pé (o menor dedo ou “o dedinho” do pé) também é uma deformidade congênita muito comum.
Também pode ser descrita como Underllapping ou 5º dedo Infra-Aduto, isto é, desviado para baixo e para dentro.
Caracteriza-se pela rotação e desvio medial das falanges do 5º dedo e contratura do tendão flexor longo, assumindo um posicionamento por baixo do 4º dedo.
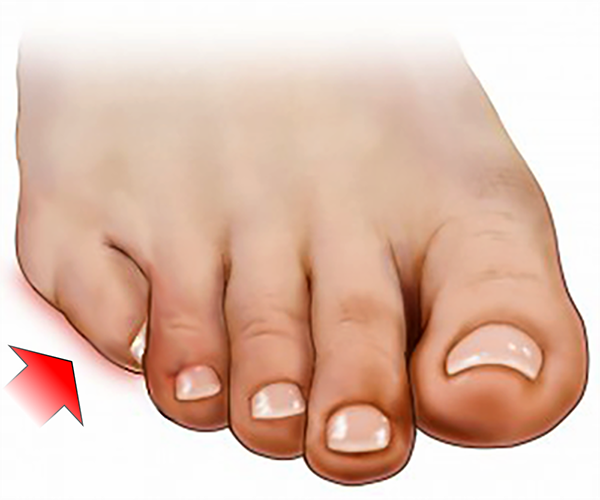
Essa deformidade causa incômodo pela compressão do 5º dedo, que fica entre o 4º dedo e o chão. A dor pode se intensificar com o uso de calçados de caixa muito baixa ou rasteiros.
Muitas vezes ocorre a formação de um calo entre o 4º e o 5º dedos (calo em espelho). A unha pode sofrer importante alteração na sua forma e no crescimento.
O tratamento conservador baseia-se na proteção com coxins (pads) para acolchoamento e mudança do estilo de calçados.
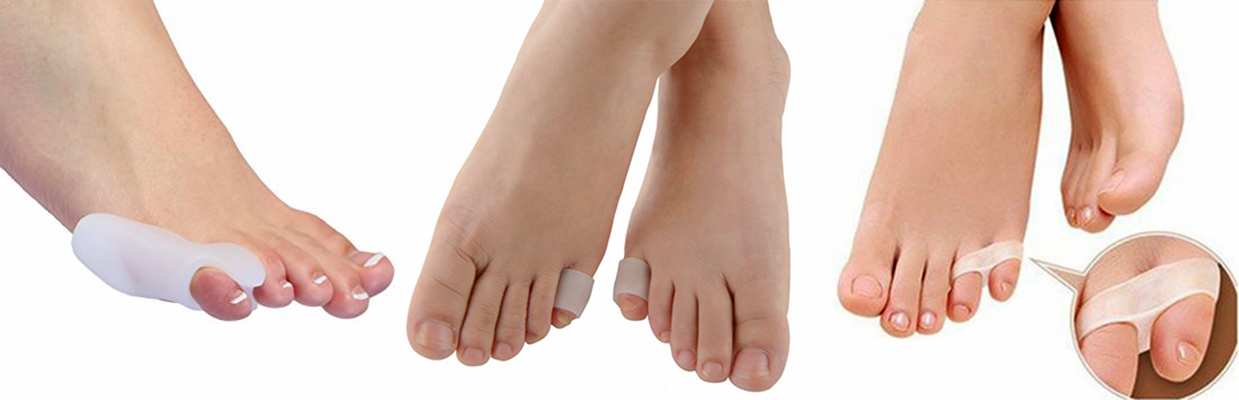
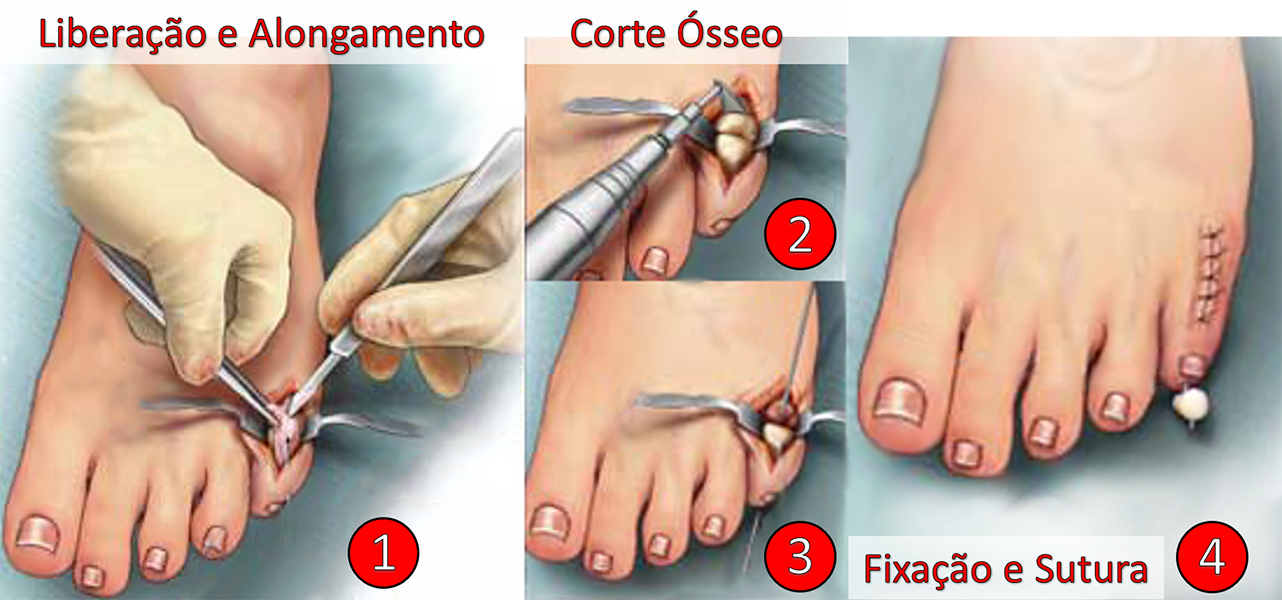
O tratamento cirúrgico consiste no alongamento e liberação das estruturas que estão encurtadas e ressecção parcial ou total da falange proximal para permitir o realinhamento do dedo.
Sindactilia e Polidactilia
Os termos SINDACTILIA e POLIDACTILIA origina-se dos prefixos gregos “SYN- “ (UNIDO, JUNTO) e “POLI-“ (MUITOS), com palavra grega “DAKTYLOS” (DEDOS, DIGITOS).
Syndaktylos = Sindactilia = Dedos Unidos
Polidaktylos = Polidactilia = Muitos Dedos
Polisyndaktilos = Polisindactilia = Muitos Dedos Unidos
SINDACTILIA
A sindactilia é a união, formada através de uma membrana de tecido mole (pele e subcutâneo) ou pela fusão óssea, entre dois ou mais dedos.
É uma alteração congênita adquirida e pode estar associada com outras alterações geneticamente determinadas como bandas de constrição (Streeter) ou síndromes como Apert, Poland, Orodigitofacial, Cornelia de Lange, Smith-Lemli-Optiz e trissomias.
A incidência da sindactilia é, em média de 1 para cada 2000 nascimentos, sendo mais comuns em homens caucasianos.
Essa deformidade pode ser classificada em:
TIPO 1 - ZIGOSINDACTILIA - União parcial ou total entre dois ou mais dedos normais, sendo mais comum entre o segundo e o terceiro dedos. É o tipo mais comum de sindactilia.
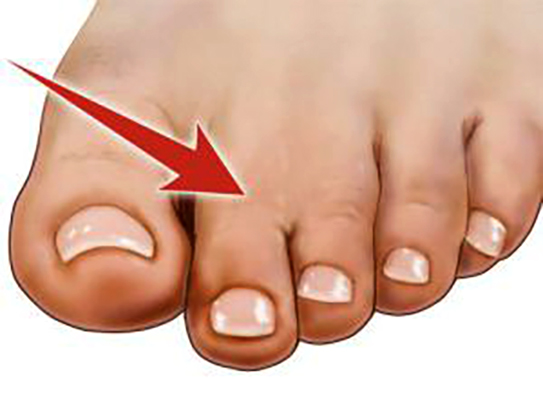
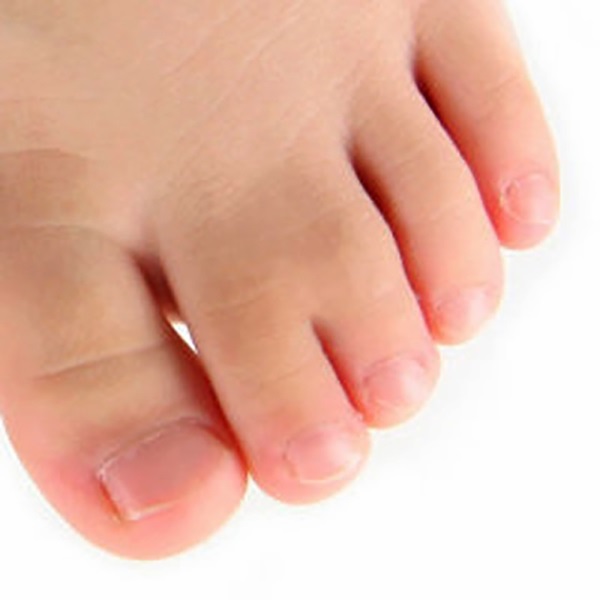
TIPO 2 - POLISINDACTILIA - União parcial ou total entre os dedos, com presença de, pelo menos, um dedo acessório (polidactilia + sindactilia). Mais comum entre o quarto ou quinto dedo e o dedo acessório.
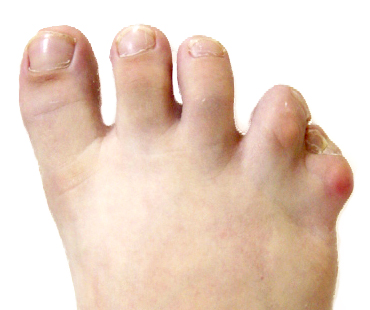
TIPO 3 – COMPLETA – Sindactilia envolvendo todos os dedos. (Síndrome de Apert)
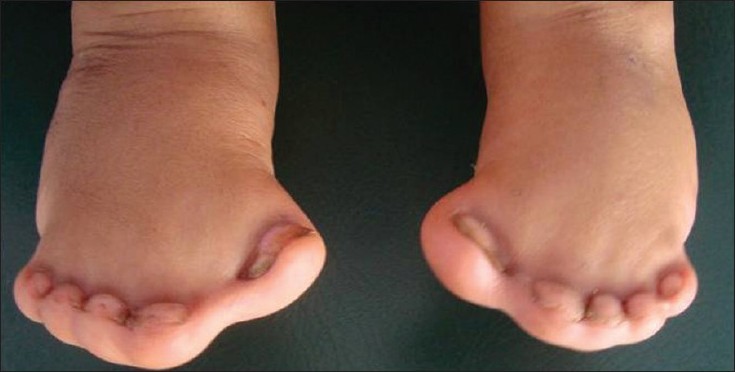
TIPO 4 – SINOSTÓTICA – Sindactilia com presença de fusão óssea (Sinostose) metatarsal
A união completa ou incompleta entre dois dedos normais do pé (zigosindactilia) raramente causa sintomas ou necessidade de correção cirúrgica.
O tratamento da polisindactilia tem o objetivo de corrigir esteticamente e/ou estreitar a largura do pé permitindo melhor acomodação em calçados fechados. Procedimentos cirúrgicos mais complexos podem ser necessários caso haja envolvimento de muitos dedos ou fusões ósseas associadas.
POLIDACTILIA
Polidactilia é aumento do número de dedos nos pés e/ou nas mãos. É uma deformidade congênita de caráter autossômico dominante e de apresentação variável, isto é, pode ocorrer de várias formas e com chance de 50 % caso um dos pais apresente essa alteração.
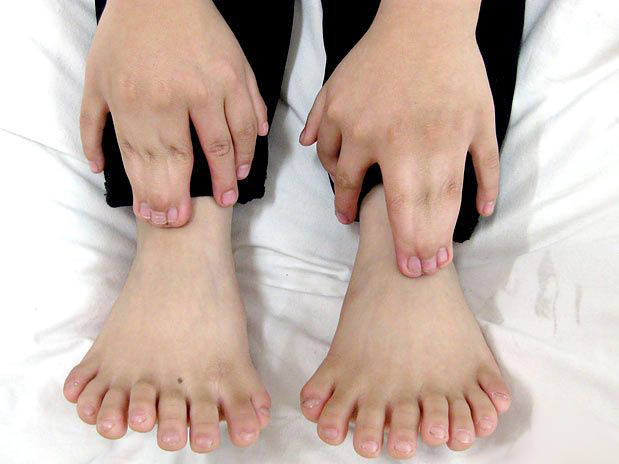
A polidactilia é comum nos pés, com incidência de 1 caso para cada 500 nascimentos; e pode associar-se com sindactilia (polisindactilia), outras malformações maiores dos membros ou síndromes geneticamente determinadas.
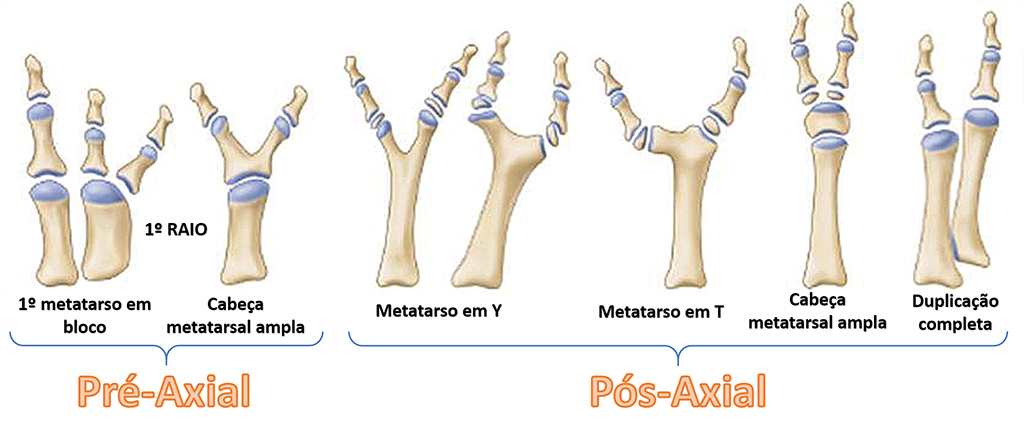
A polidactilia pode ser classificada como:
PRÉ-AXIAL - Acomete o primeiro dedo (dedão) do pé - 80% dos casos
CENTRAL - Acomete o segundo, terceiro e/ou quarto dedos - 6% dos casos
PÓS-AXIAL - Acomete o quinto dedo - 14% dos casos
Outra classificação (Venn-Watson) divide as deformidades somente em pré-axiais e pós-axiais, sendo subdivididas em 6 subcategorias:
O tratamento cirúrgico normalmente está indicado por razões estéticas ou para permitir o uso de calçados e consiste em remover o(s) dedo(s) acessório(s). Ocasionalmente cirurgias mais complexas são necessárias quando a polidactilia está associada à sindactilia (polisindactilia).
Gigantismo e Macrodactilia
É uma deformidade congênita rara. Caracteriza-se pelo crescimento excessivo de um ou mais dedos do pé. Quando acomete toda a extensão do pé ou da mão, ou até mesmo todo o membro, é denominada de gigantismo.
Gigantismo é também a denominação do hipercrescimento corporal ocasionado pela produção excessiva de hormônio do crescimento (GH) durante a infância ou adolescência, e caracteriza-se pelo aumento descomunal da estatura corporal. Quando o excesso de produção hormonal ocorre na idade adulta chamamos a alteração de acromegalia, que afeta principalmente o crescimento das cartilagens e da largura dos ossos, não alterando a altura do indivíduo.
Frequentemente a deformidade aumenta com o crescimento e está associada com hiperplasia congênita do tecido linfático ou adiposo (macrolipodistrofia). Doenças como neurofibromatose e síndromes congênitas podem estar associadas à presença de gigantismo ou macrodactilia nas mãos e/ou nos pés.
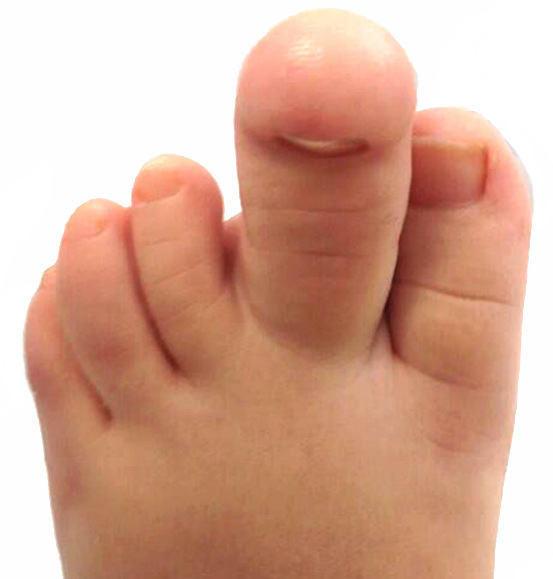
A macrodactilia isolada é causada pela macrolipodistrofia, crescimento anormal do tecido gorduroso e ósseo. Normalmente é unilateral, afetando mais o segundo ou o terceiro dedo do pé e não apresentando outras anomalias concomitantes.
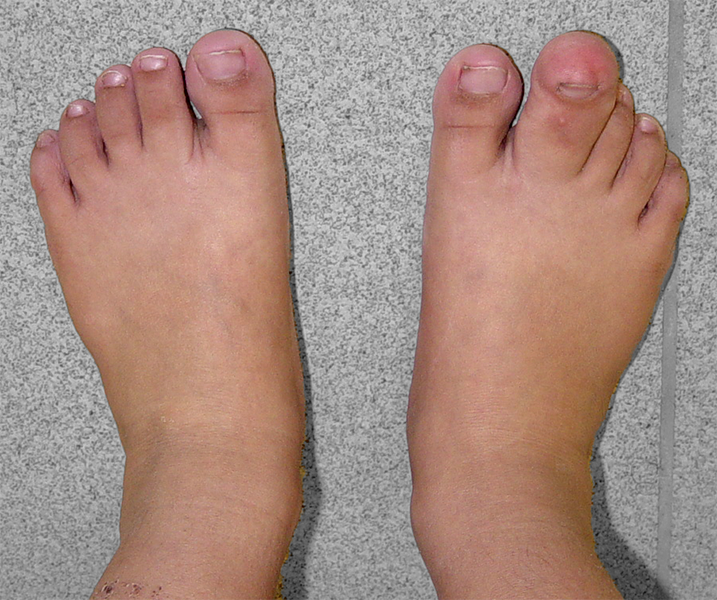
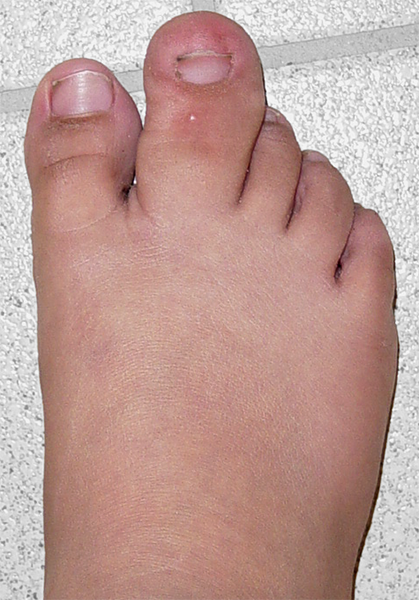
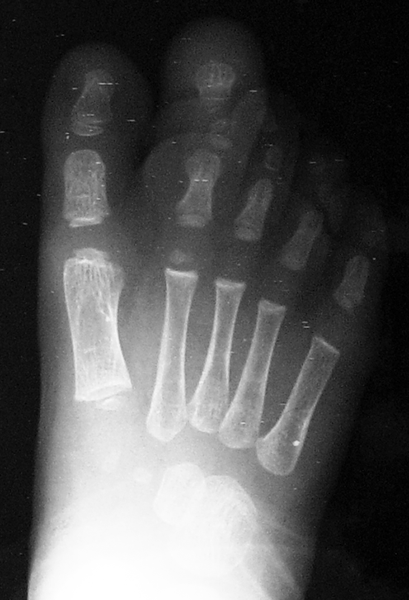
A lipofibromatose congênita ocasiona o aumento localizado somente do tecido gorduroso e ligamentar, sem alteração do crescimento ósseo, podendo acometer as palmas das mãos e/ou as plantas dos pés.
A macrodactilia associada a neurofibromatose (Doença de Von Recklinghausen) é rara, normalmente unilateral e associada a formação de um neurofibroma plexiforme.
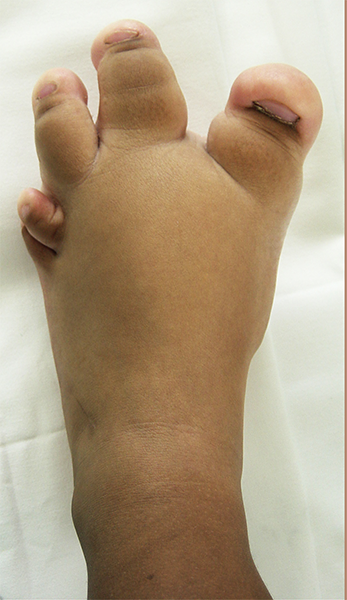
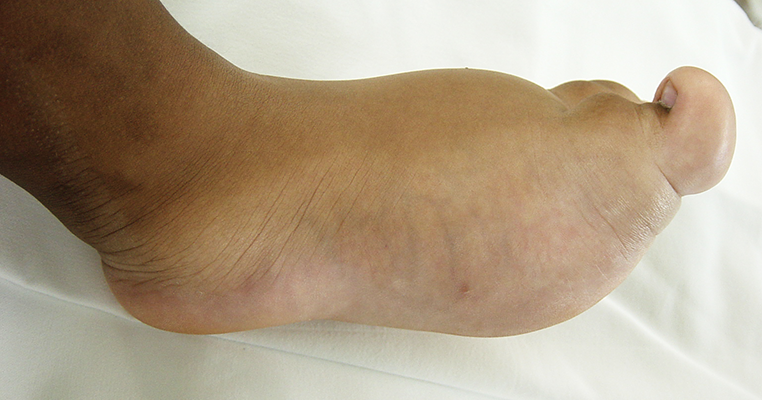
Neurofibromatose - Doença de Von Recklinghausen
A síndrome de Proteus é uma alteração genética raríssima, não hereditária, podendo se apresentar de múltiplas formas, com tumorações subcutâneas, alterações e deformidades ósseas e com gigantismo dos membros, principalmente pernas e pés.
O caso de maior repercussão é o de Joseph Merrick, o homem-elefante, o qual inspirou o filme de 1980 que conta a sua história. Joseph tinha 90% de seu corpo deformado pela doença. Atualmente, acredita-se que o homem-elefante possuía a síndrome de Proteus, possivelmente associada à neurofibromatose.
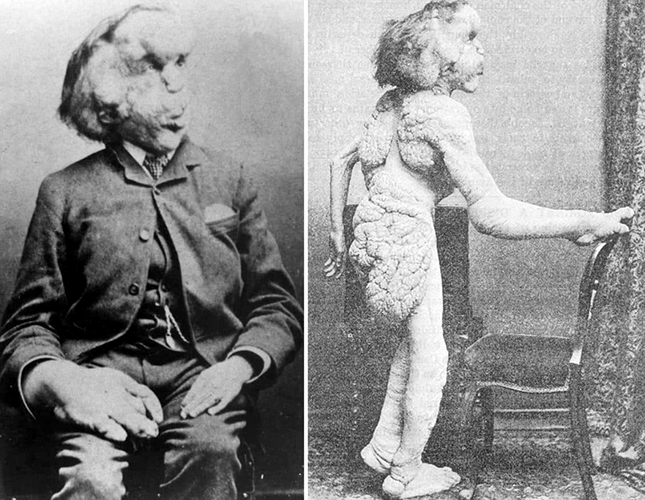
Joseph Merrik (1862-1890) - O Homem-Elefante (Inglaterra)
Existem pouco mais de 100 casos no mundo de síndrome de Proteus, grande parte apresenta gigantismo dos membros inferiores e pés.
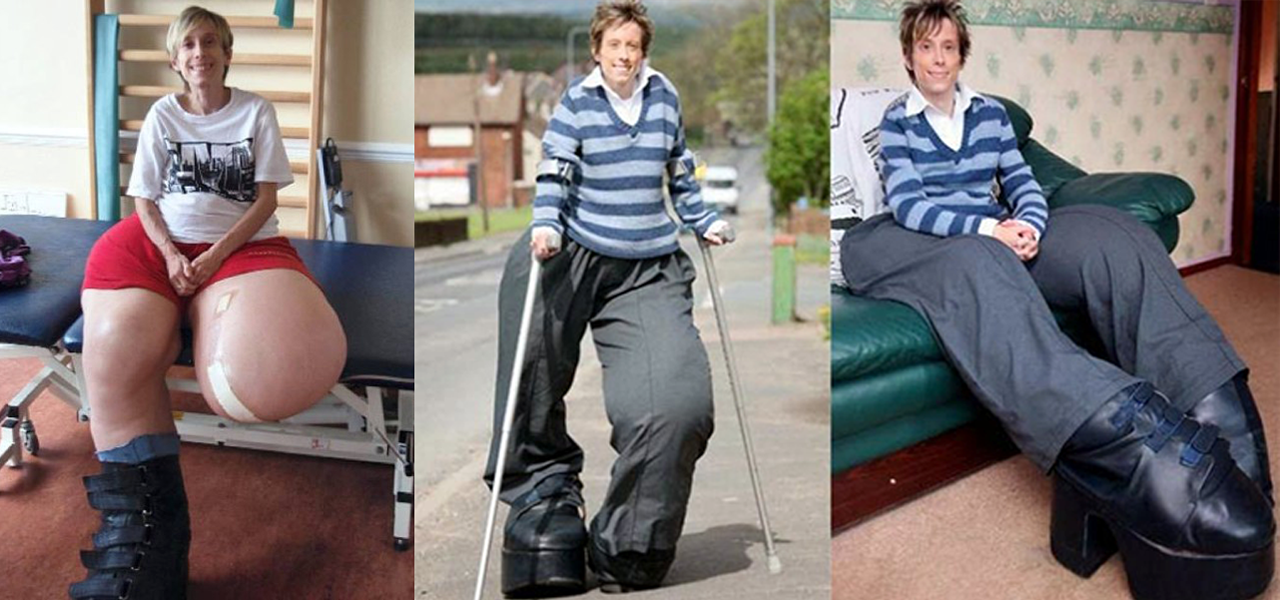
S. de Proteus - Mandy Sellars (Inglaterra)

S. de Proteus - Verdant Joshi (Índia)
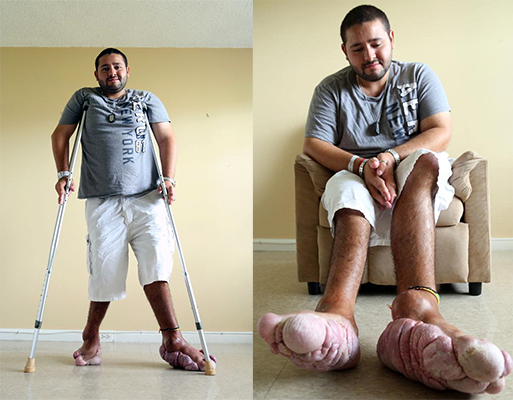
S. de Proteus - Jeffrey Ortega (USA)
A hipertrofia de partes moles com crescimento ósseo anormal está presente na síndrome de Klippel-Trenaunay (angio-osteo-hipertrofia ou hipertrofia hemangiectática). Na maioria dos casos o acometimento é unilateral e atinge todo o membro, podendo apresentar alterações vasculares como hemagiomas e veias varicosas.
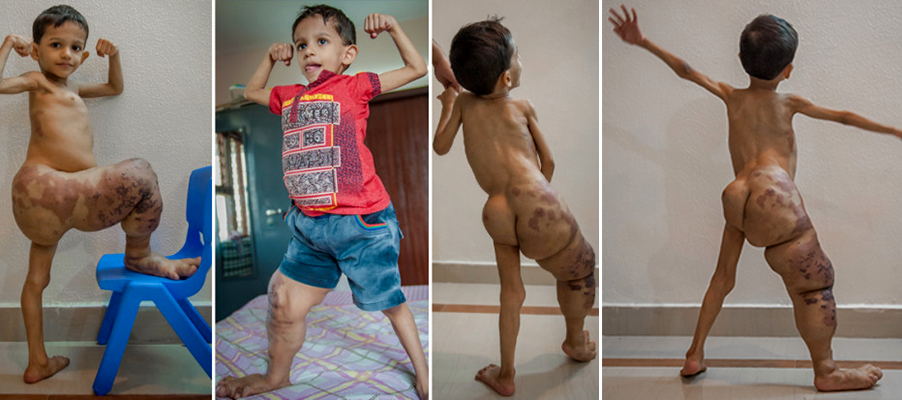
S. de Klippel-Trenaunay - Akshaj Khandelwal (Índia)
A indicação cirúrgica tem por objetivo melhorar a aparência estética, permitir um melhor apoio do pé e possibilitar o uso de calçados fechados. Muitas vezes é necessária a amputação parcial ou total do dedo ou do membro afetado. A médio e longo prazo pode ocorrer a recidiva da deformidade e/ou a necessidade de cirurgias reparadoras adicionais.
Pé Fendido
A deformidade em pé fendido (Split Hand/Foot Malformation (SHFM) ou Lobster Foot (Pé de Lagosta)) é cientificamente chamada de Ectrodactilia, uma anomalia congênita do desenvolvimento embriológico, de herança autossômica dominante com penetrância variável, isto é, pode aparecer de várias formas com uma chance de 50% se um dos pais tiver a alteração.
Na maioria das vezes acomete ambos os pés e eventualmente ambas as mãos podem também apresentar a mesma alteração.
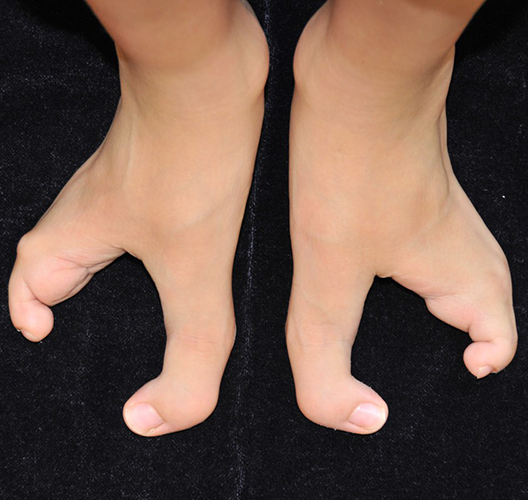
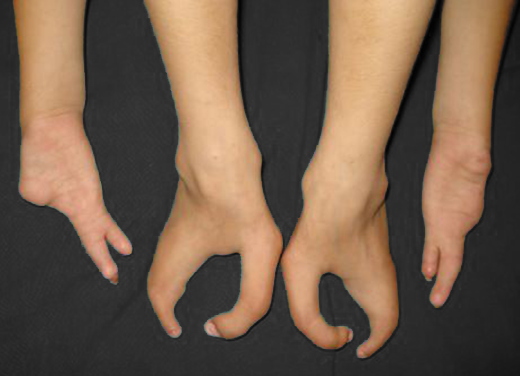
É uma deformidade congênita muito rara (1:90.000) que afeta mais o sexo masculino.
Sua apresentação é caracterizada por uma divisão medial (fenda) e ausência parcial ou completa dos dedos centrais, podendo acometer os pés e/ou as mãos e aparecer isoladamente ou associada à outras síndromes congênitas. Os dedos presentes podem estar unidos ou interligados lateralmente pela pele (sindactilia) e ter ausência ou atrofia (hipoplasia) de falanges.
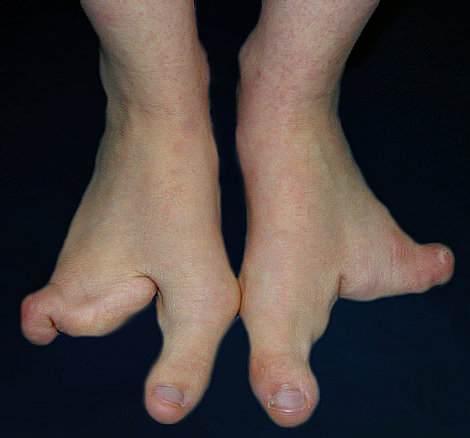
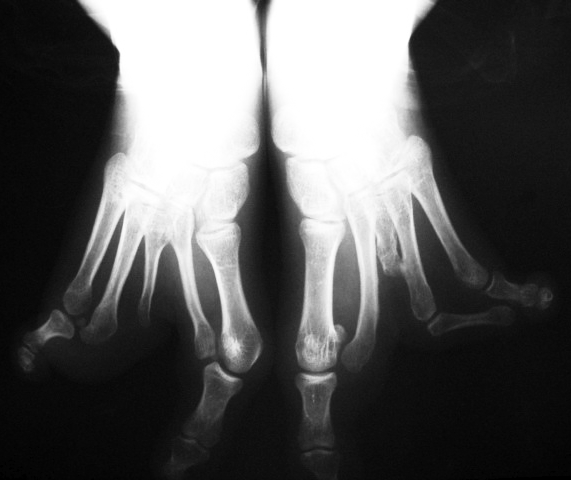
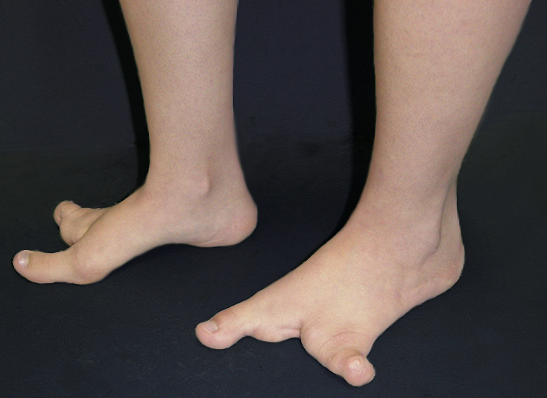
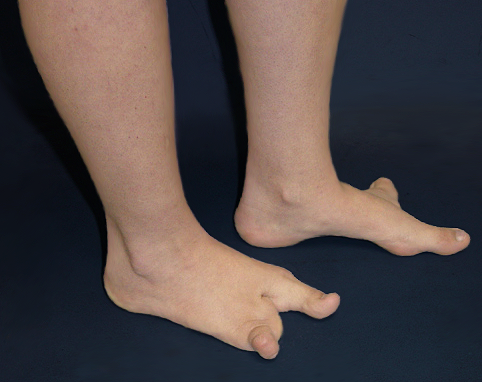
O tratamento consiste em conseguir dar um melhor alinhamento e favorecer a função do membro afetado. Nos pés, o objetivo principal é fornecer ao paciente a possiblidade do uso de calçados normais, evitando zonas de atrito e calosidade. Para isso, muitas vezes é necessário o uso de protetores, bandagens para amarração/contenção e confecção de calçados especiais.
A intervenção cirúrgica é indicada para as deformidades mais graves, quando existe grande dificuldade de adequar calçados ou órteses, e tem por finalidade propiciar um melhor alinhamento dos dedos e o fechamento parcial ou total da fenda, buscando a maior funcionalidade possível do membro.
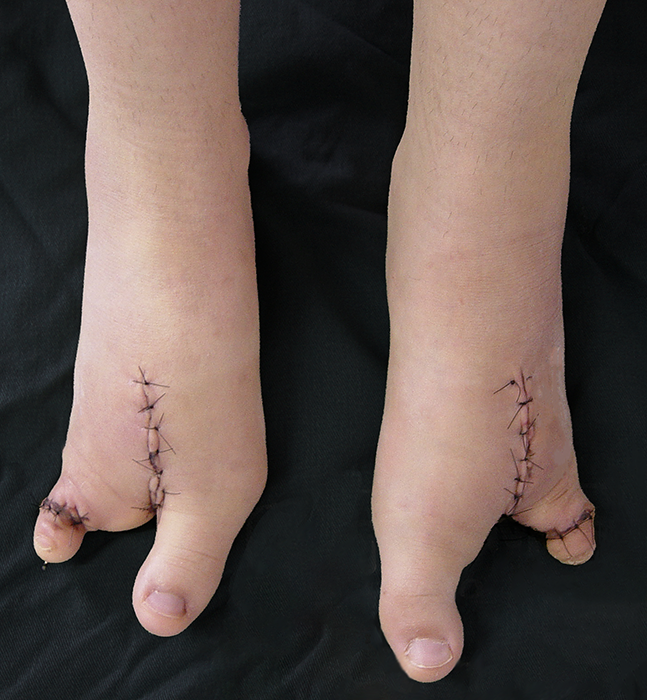
Devido à herança autossômica, a prevenção da malformação é o esclarecimento genético dos riscos, associado ao diagnóstico precoce ultrassonográfico no pré-natal.
Charcot-Marie-Tooth
A doença de Charcot-Marie-Tooth (CMT) é a alteração hereditária mais comum que acomete os nervos periféricos.
Também é denominada de Neuropatia Motora-Sensorial Hereditária ou Atrofia Muscular Fibular.
A doença de Charcot-Marie-Tooth foi descrita por três médicos em 1886: Jean Martin Charcot e Pierre Marie de Paris, França, e Howard Henry Tooth de Cambridge, Inglaterra.

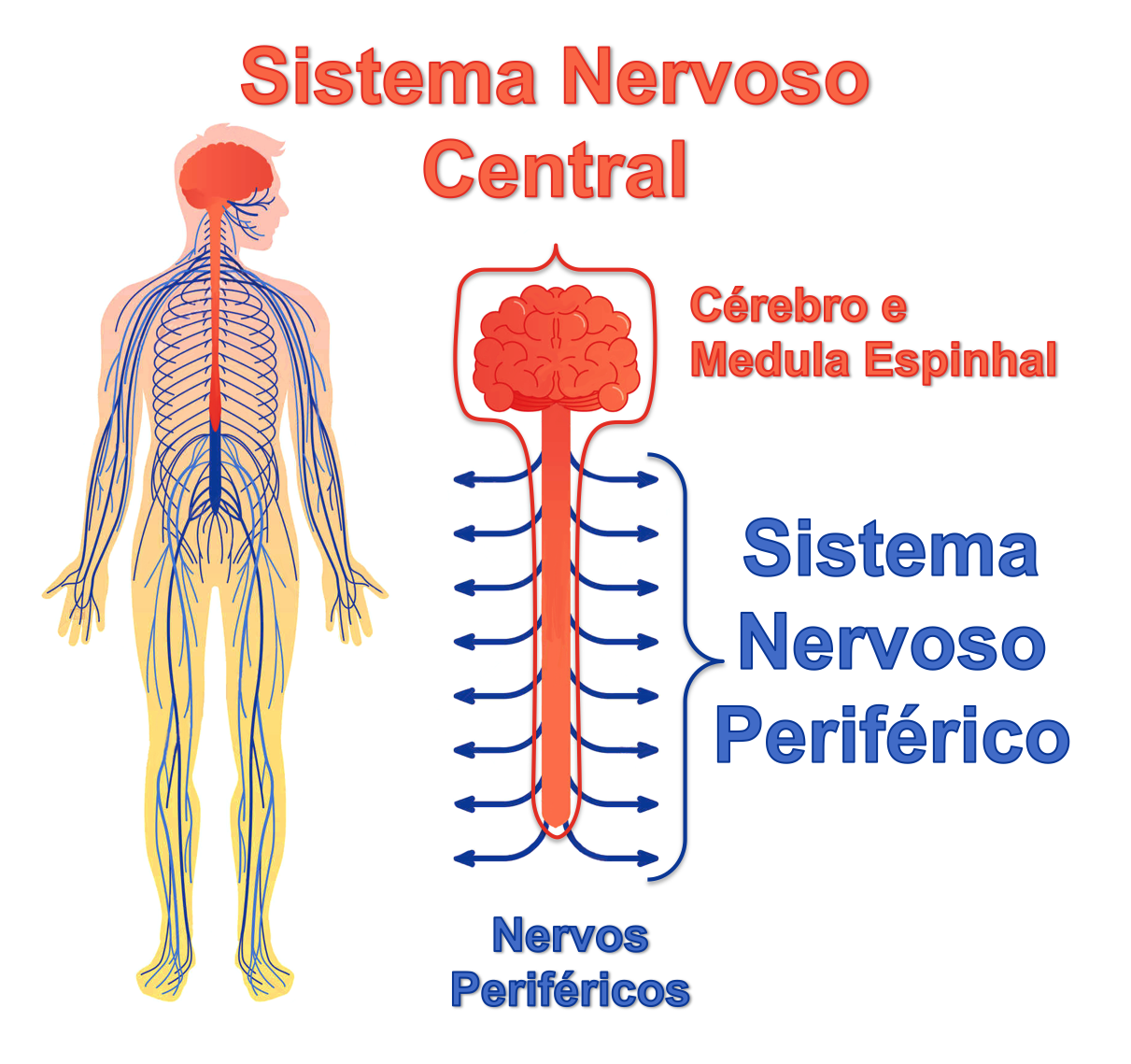
O conjunto de ramificações nervosas (nervos) que saem da medula espinhal é chamado de sistema nervoso periférico. Esse sistema leva os estímulos motores do cérebro e da medula espinhal para os músculos do corpo e trazem os estímulos sensitivos e proprioceptivos da pele, tendões, músculos e articulações para serem interpretados pelo sistema nervoso central (medula espinhal e cérebro).
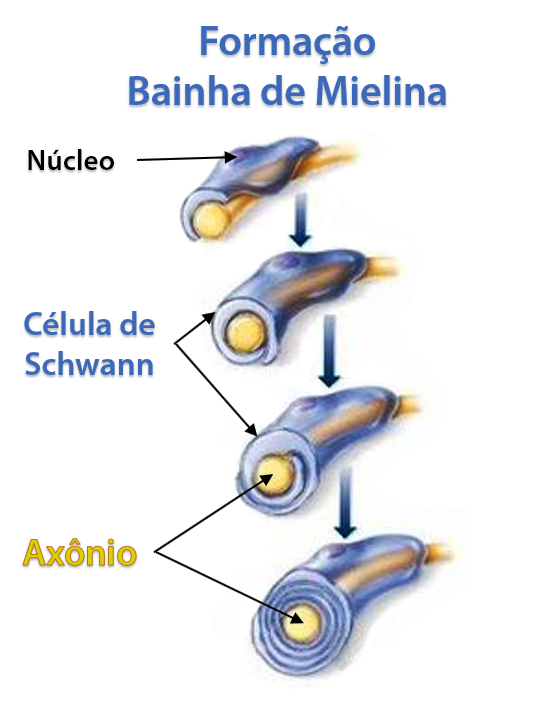
Os nervos periféricos do corpo humano são recobertos por mielina, uma bainha lipídica que protege o neurônio e acelera o impulso elétrico nervoso. As células que formam essa bainha de mielina são chamadas de Células de Schwann.
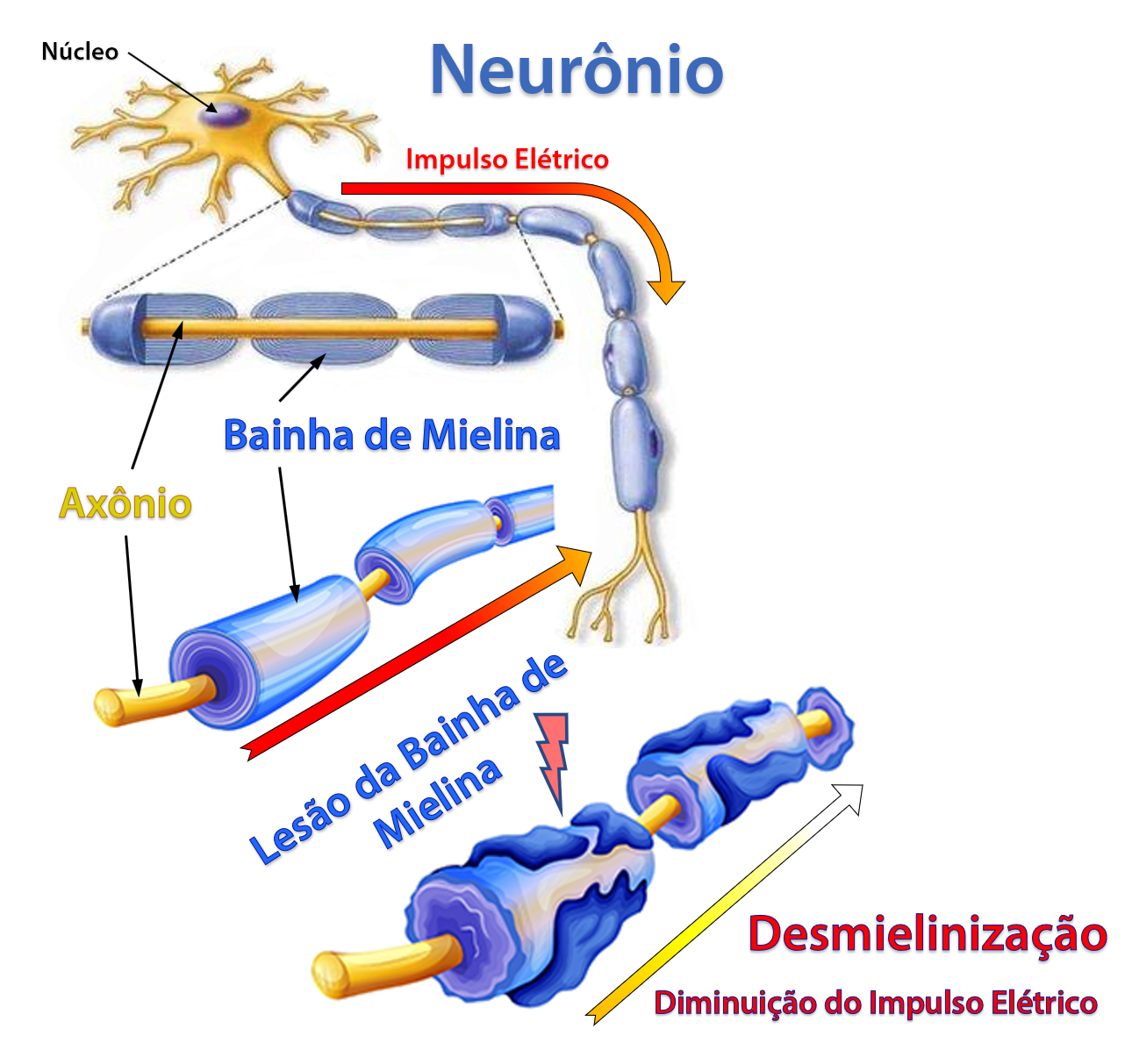
A doença de Charcot-Marie-Tooth é uma doença desmielinizante, isto é, acomete a formação da bainha de mielina que recobre as fibras axonais dos nervos periféricos, alterando a condução do estimulo nervoso e causando a degeneração axonal do neurônio. Isso altera a função normal dos nervos, diminuindo a velocidade de condução elétrica, causando perda da sensibilidade e desequilíbrio na força muscular, principalmente dos membros inferiores.
A doença de Charcot-Marie-Tooth (CMT) pode ser herdada através de vários padrões genéticos e acometer diferentes partes do DNA herdado (alterações cromossômicas); por isso, ela apresenta diversas formas clínicas, sendo subdividida em diversos tipos:
CMT1 – É a forma mais comum (70 % dos pacientes com CMT), de padrão genético autossômico dominante (gene anormal pode ser herdado de qualquer um dos pais (50% de chance de herdar e 50 % de chance de desenvolver a doença)). Caracteriza-as pela perda gradativa da velocidade de condução elétrica dos nervos a partir da segunda década de vida (adolescentes e adultos jovens). A CMT1 pode ainda ser subdividida de acordo com o gene acometido: CMT1-A – Alteração no cromossomo 17, CMT1-B – Alteração no cromossomo 1 e CMT1-C – Alteração no cromossomo 16.
CMT2 - É a forma menos pronunciada e de acometimento tardio, normalmente após a quarta ou quinta década de vida. Altera principalmente as proteínas do axônio e atinge menos a bainha de mielina, causando menor perda motora e maior perda sensitiva. A CMT2 também pode ser subdividida de acordo com o gene acometido: CMT2-A – Alteração no cromossomo 1, CMT2-B – Alteração no cromossomo 3, CMT2-C – Alteração no cromossomo 12 e CMT2-D – Alteração no cromossomo 7.
CMT3 – Doença de Dejerine-Sottas. Forma rara e agressiva de degeneração da bainha de mielina que inicia na infância e causa importante perda motora e sensitiva. Crianças acometidas apresentam atrofia muscular severa, fraqueza e perda importante da sensibilidade.
CMT4 – Compreende diversos subtipos de neuropatias desmielinizantes de padrão autossômico recessivo (gene deve ser herdado dos dois pais para apresentar a doença). São raras, com sintomas progressivos que iniciam normalmente na infância.
CMTX – Doença ligada à alteração do cromossomo X, que altera a forma da bainha de mielina. Os homens apresentam formas mais graves da doença ao herdar o cromossomo X defeituoso de suas mães.
A suspeita diagnóstica ocorre através da história familiar e da avaliação clínica ortopédica e neurológica. O exame de neurocondução e eletromiografia dos membros inferiores testa a velocidade de condução e integridade dos nervos das pernas e dos pés. Exames de avaliação genética confirmam o diagnóstico com a identificação do defeito cromossômico no DNA do paciente (testes de mutação CMT). Caso os testes e exames forem inconclusivos, pode ser feita a biópsia de nervo, a retirada de um pequeno fragmento de um nervo da perna para estudo microscópico das células neuronais e da bainha de mielina (células de Schwann).
Os sintomas da principal forma da doença de Charcot-Marie-Tooth (CMT1) iniciam na infância ou adolescência e progridem com o tempo, mas em outras apresentações (CMT2) os sintomas podem iniciar na idade adulta.
Na maioria das vezes o desequilíbrio neuromuscular progride lentamente dos pés e pernas até os braços e mãos. A gravidade e apresentação dos sintomas variam enormemente, mesmo entre pessoas afetadas da mesma família. Embora existam raros casos graves, onde a doença afeta os músculos da respiração, a doença de Charcot-Marie-Tooth não é considerada uma doença fatal; e a maioria dos pacientes conseguem ter uma expectativa de vida normal.
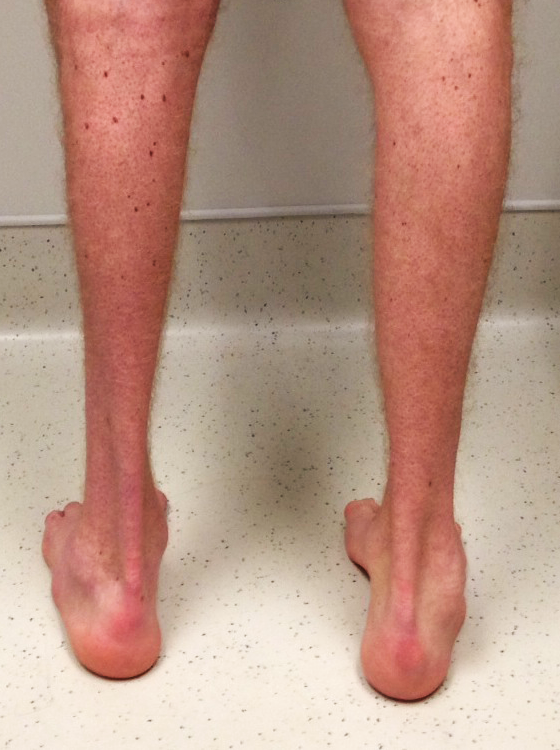
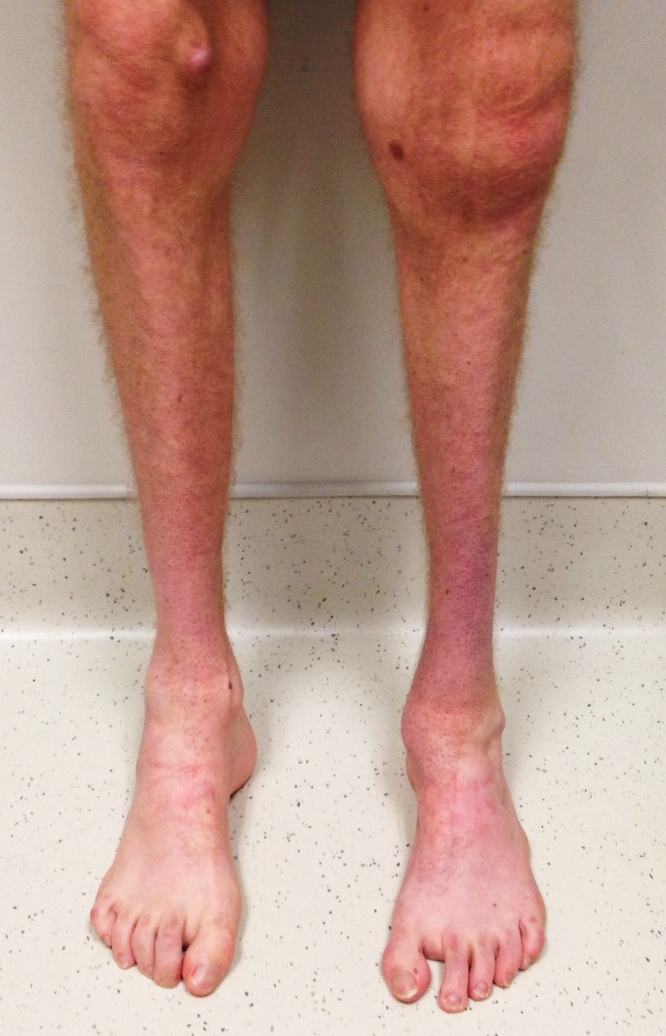
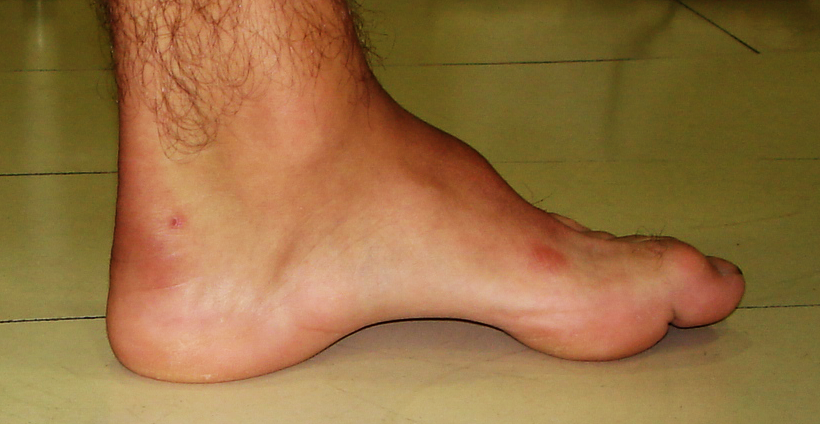
Os principais sintomas e deformidades associadas à Doença de Charcot-Marie-Tooth são:
- Fraqueza e perda muscular nas mãos, pernas, tornozelos e pés
- Encurtamento muscular da panturrilha (Pé Equino)
- Aumento do arco dos pés (pés cavos)
- Deformidade dos dedos dos pés e mãos (Dedos em Garra)
- Alteração da marcha, tropeços e quedas frequentes ao caminhar
- Dificuldade para correr
- Diminuição da sensibilidade nos pés e nas pernas
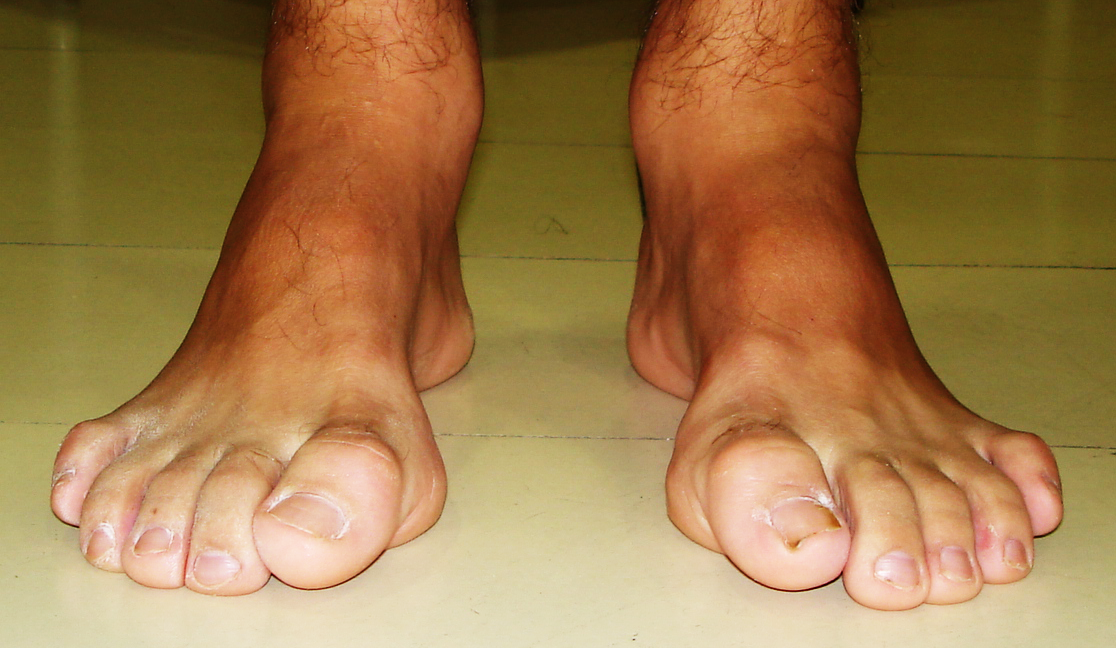
O tratamento da doença de Charcot-Marie-Tooth baseia-se na orientação e suporte ao paciente. O acompanhamento multidisciplinar com ortopedista, neurologista e fisioterapeuta visa atenuar os efeitos progressivos da doença. O uso de órteses, para alinhar e estabilizar as deformidades do pé e da perna, assim como procedimentos cirúrgicos de alongamentos ou transferências de tendão são frequentemente indicados. A fisioterapia e a terapia ocupacional contínua deve ser instituída para melhorar e proporcionar uma qualidade de vida duradoura.
Não existe cura para a doença de Charcot-Marie-Tooth. No entanto, considerando sua progressão lenta, com o bom entendimento e o suporte médico adequado pode-se conseguir alívio sintomático e almejar uma expectativa de vida normal.
Mais informações relevantes podem ser encontradas no site da Associação Brasileira dos Portadores de Charcot-Marie-Tooth.

Pé Torto Congênito
